Alineamiento de secuencias¶
Significado biológico¶
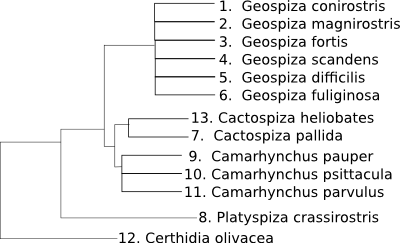
Las secuencias de ADN y proteína definen la función de las proteínas en los seres vivos.
Cuando más similares sean dos secuencias más similares tenderán a ser las funciones de las proteínas codificadas por ellas.
Las secuencias de un mismo gen en un conjunto de especies serán más distintas cuanto más alejadas filogenéticamente estén las especies comparadas.

(From BSCS, Biological Science: Molecules to Man, Houghton Mifflin Co., 1963).
Normalmente dos secuencias tienen una alta similitud porque son homólogas, es decir comparten un ancestro común.
A diferencia de la similitud, la homología no es un término cuantitativo, dos secuencias o son homólogas, derivan del mismo ancestro, o no lo son.
A partir de la similitud de las secuencias inferimos la homología.
La acumulación de mutaciones en el ADN a lo largo del tiempo es la causa de que las secuencias de un mismo gen en dos especies distintas no sean idénticas.
Cuanto más tiempo pase desde el último antecesor común más diferentes serán las secuencias.
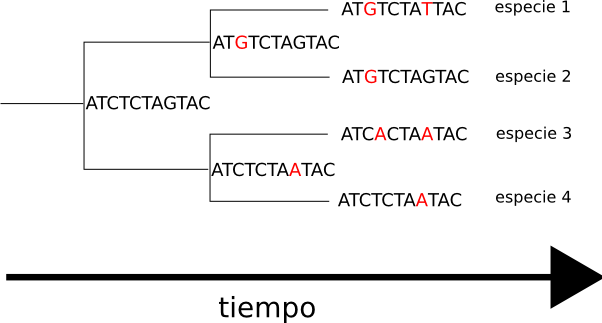
En las secuencias, además de sustituciones de un residuo por otro pueden darse inserciones o deleciones.
Utilidad¶
Los alineamientos sirven, entre otras cosas para:
Asegurarse de que dos secuencias son similares y cunatificar su similitud.
Encontrar dominios funcionales.
Comparar un gen y su producto.
Buscar posiciones homólogas en las secuencias.
Alineamiento de secuencias¶
Para poder cuantificar el grado de similitud de dos secuencias lo primero que hay que hacer es alinearlas.
Sin alinear
CGATGCTAGCGTATCGTAGTCTATCGTAC
| ||
ACGATGCTAGCGTTTCGTATCATCGTA
Alineadas
-CGATGCTAGCGTATCGTAGTCTATCGTAC
|||||||||||| |||||||||||||||
ACGATGCTAGCGTTTCGTA-TC-ATCGTA-
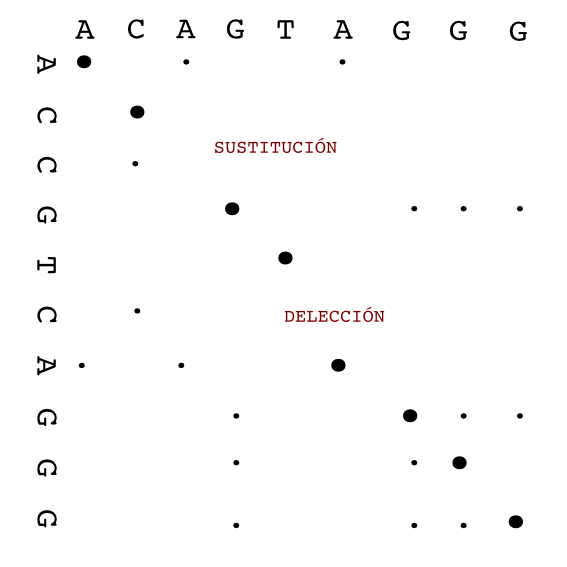
Una sustitución es un cambio de un residuo por otro.
Cuando falta una base decimos que hay un gap.
Un gap puede corresponder tanto a una deleción como a una inserción.
Debido a la existencia de gaps y sustituciones alinear dos secuencia no es trivial.
Pueden existir diferentes alineamientos dependiendo del número de gaps que permitamos introducir.
Sin gaps (10 coincidencias):
a: ATATTGCTACGTATATCAT
||||||||||
b: ATATATGCTACGTATCAT
Con gaps en a (14 coincidencias):
a: ATAT-TGCTACGTATATCAT
|||| ||||||||||
b: ATATATGCTACGTATCAT
Con gaps en a y b (16 coincidencias):
a: ATAT-TGCTACGTATATCAT
|||| ||||||| ||||||
b: ATATATGCTACG--TATCAT
El objetivo del alineamiento es conseguir alinear las posiciones homólogas.
Puntuación de los alineamientos¶
El alineamiento con mejor puntuación debería ser el más razonable desde un punto de vista biológico, el que alinea más posiciones homólogas.
Se recurre a medidas objetivas.
Para comparar distintos alineamientos entre sí se pueden asignar puntuaciones.
Ejemplos de sistemas de puntuación:
Número de letras que coinciden
Porcentaje de identidad, número de coincidencias cada cien posiciones.
Porcentaje de similitud, tiene en cuenta la similitud fisicoquímica de los diferentes aminoácidos.
En la práctica se suelen utilizar sistemas de puntuación más complejos que también tienen en cuenta los gaps.
Se suelen incluir dos penalizaciones para los gaps, una para abrir el gap y otra para extenderlo. Este último suele ser menos costoso.
De entre todos los alineamientos posibles el óptimo es el que presenta una máxima puntuación para el sistema de puntuación dado.
Ejemplo de puntuación de un alineamiento.
Sistema de puntuación: match: +1, mismatch: 0, gap: -1
G-ATESLIKESCHEESE
| ||| ||||||
GRATED-----CHEESE
puntuación: 10 matches * 1 + 1 mimatch * 0 + 6 gaps * -1 = 4
Para buscar el alineamiento óptimo se han desarrollado varios algoritmos y programas.
Alineamientos globales y locales¶
Hay dos tipos de alineamientos principales: globales y locales.
En el global se intenta que el alineamiento cubra las dos secuencias completamente introduciendo los gaps que sean necesarios.
En el local se alinean sólo las zonas más parecidas.
El gobal sirve para alinear secuencias que se empiecen y acaben en la misma región, por ejemplo genes homólogos de especies similares.
El alineamiento local suele ser la mejor opción a no ser que se esté seguro de que las los secuencias deben de parecerse a lo largo de toda sus extensión. En muchos casos las secuencias homólogas se parecen sólo en las regiones más conservadas.
Local
TAGCTA-TCGTAG
|||||| ||||||
TAGCTAGTCGTAG
Global
TACGGGGCTAGCTA-TCGTAG
|||| ||| ||||||
TAGC----TAG----TCGTAG
El método a elegir dependerá del objetivo. Se puede hacer un alineamiento para ver si dos proteínas tienen dominios similares, si pertenecen a la misma familia, si tienen un antecesor común.
Entre otras decisiones que hay que tomar hay que decidir si queremos hacer un alineamiento global o local, la matriz de puntuación (scoring matrix), el valor de la penalización por gaps o por no coincidencias (mismatches).
Los programas vienen con valores por defectos que han sido pensados para resolver de forma óptima un problema concreto, si ese es nuestro problema esos son los mejores valores para los parámetros. Si no es así debemos preocuparnos por entender como funciona el programa y como afectan los distintos parámetros.
Es muy fácil hacer un alineamiento cuando las secuencias son muy parecidas, pero a medida que se diferencian más el asunto se complica. El límite para las secuencias de proteínas está alrededor del 25% de identidad.
Métodos de alineamiento¶
Matriz de puntos (dot matrix o dot plot)¶
Representa los alineamientos gráficamente.
Da unos resultados intuitivos e informa de posibles alineamientos alternativos.
Sirve para descubrir repeticiones, inserciones, deleciones (largas).
También se puede utilizar para identificar regiones similares que después podemos alinear por otro método.
Una ventaja y a la vez una desventaja es que el resultado es gráfico. En la horizontal del gráfico se representa una secuencia y en la vertical otra.
El algoritmo consiste en recorrer la secuencia A base por base (o utilizando una ventana de varias bases). Para cada posición de la secuencia A se estudian todas las posiciones de la secuencia B, si las posiciones son iguales se pone un punto, si son distintas no se pone.
Los segmentos de secuencia similar se detectan como diagonales.
El número de letras de las palabras (words) determina la sensibilidad.
Se suelen aplicar filtros para mejorar el gráfico eliminando parecidos espúreos.
Normalmente no se comparan las secuencias letra por letra sino grupos de bases (words).
Se comparan todas las palabras (words) de una secuencia con las de la otra y se dibuja una pequeña diagonal cuando son suficientemente parecidas.
Para determinar si dos palabras son suficientemente similares se aplican los métodos de puntuación que veremos después.
>P05049|SNAK_DROME Serine protease snake - Drosophila melanogaster (Fruit fly).
MIILWSLIVHLQLTCLHLILQTPNLEALDALEIINYQTTKYTIPEVWKEQPVATIGEDVD
DQDTEDEESYLKFGDDAEVRTSVSEGLHEGAFCRRSFDGRSGYCILAYQCLHVIREYRVH
GTRIDICTHRNNVPVICCPLADKHVLAQRISATKCQEYNAAARRLHLTDTGRTFSGKQCV
PSVPLIVGGTPTRHGLFPHMAALGWTQGSGSKDQDIKWGCGGALVSELYVLTAAHCATSG
SKPPDMVRLGARQLNETSATQQDIKILIIVLHPKYRSSAYYHDIALLKLTRRVKFSEQVR
PACLWQLPELQIPTVVAAGWGRTEFLGAKSNALRQVDLDVVPQMTCKQIYRKERRLPRGI
IEGQFCAGYLPGGRDTCQGDSGGPIHALLPEYNCVAFVVGITSFGKFCAAPNAPGVYTRL
YSYLDWIEKIAFKQH
>gi|129688|sp|P21902|PCE_TACTR Proclotting enzyme precursor
MLVNNVFSLLCFPLLMSVVRCSTLSRQRRQFVFPDEEELCSNRFTEEGTCKNVLDCRILLQKNDYNLLKE
SICGFEGITPKVCCPKSSHVISSTQAPPETTTTERPPKQIPPNLPEVCGIHNTTTTRIIGGREAPIGAWP
WMTAVYIKQGGIRSVQCGGALVTNRHVITASHCVVNSAGTDVMPADVFSVRLGEHNLYSTDDDSNPIDFA
VTSVKHHEHFVLATYLNDIAILTLNDTVTFTDRIRPICLPYRKLRYDDLAMRKPFITGWGTTAFNGPSSA
VLREVQLPIWEHEACRQAYEKDLNITNVYMCAGFADGGKDACQGDSGGPMMLPVKTGEFYLIGIVSFGKK
CALPGFPGVYTKVTEFLDWIAEHMV
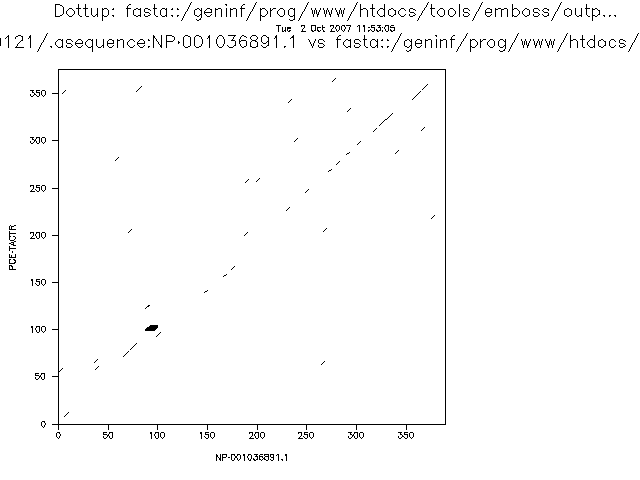
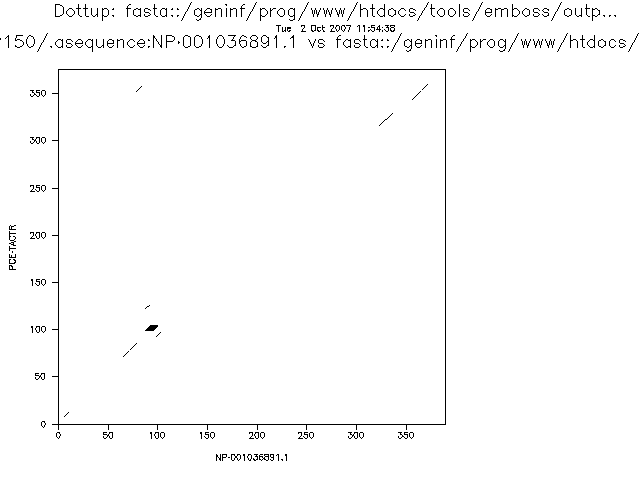
El alineamiento de estas dos secuencias se ve bien con word size 3 o 4.
Hay varios programas para aplicar este método, uno de ellos es el dotmatcher del emboss.
Es fácil detectar inserciones y deleciones grandes, repeticiones directas e invertidas.

Gen duplicado en tandem.
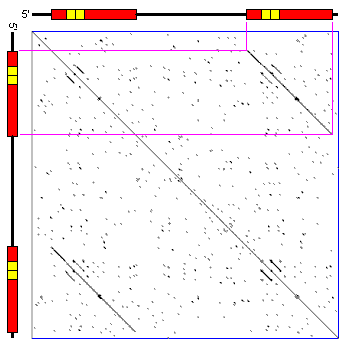
Análisis del genoma de Arabidopsis. http://www.arabidopsis.org/
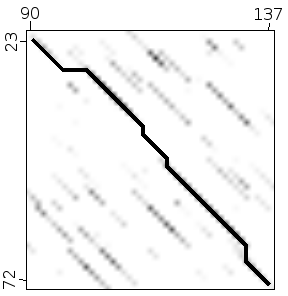
Los gráficos dotplot sugieren caminos a través del espacio de alinamientos posibles.
Estas rutas son un alinamiento.
PLAU 90 EPKKVKDHCSKHSPCQKGGTCVNMP--SGPH-CLCPQHLTGNHCQKEK---CFE 137
PLAT 23 ELHQVPSNCD----CLNGGTCVSNKYFSNIHWCNCPKKFGGQHCEIDKSKTCYE 72
El método tiene una limitación importante, nos muestra las regiones similares, pero no obtenemos un alineamiento.
Algoritmo de programación dinámica¶
Este método permite encontrar el alineamiento óptimo dadas dos secuencias y un esquema de puntuación determinado.
En principio estos alineamientos se corresponden con las diagonales del método gráfico.
Fue utilizado por primera vez por Needleman y Wunsch para hacer alineamientos globales
########################################
# Program: needle
# Rundate: Tue Oct 02 10:58:05 2007
# Align_format: srspair
# Report_file: /ebi/extserv/old-work/needle-20071002-10580428983041.output
########################################
#=======================================
#
# Aligned_sequences: 2
# 1: SNAK_DROME
# 2: PCE_TACTR
# Matrix: EBLOSUM62
# Gap_penalty: 10.0
# Extend_penalty: 0.5
#
# Length: 462
# Identity: 126/462 (27.3%)
# Similarity: 184/462 (39.8%)
# Gaps: 114/462 (24.7%)
# Score: 440.5
#
#
#=======================================
SNAK_DROME 1 MIILWSLIVH--LQLTCLHLILQTPNLEALDALEIINYQTTKYTIPEVWK 48
::|: ..|.|..|::.......| :.|..::..|
PCE_TACTR 1 MLVNNVFSLLCFPLLMSVVRCSTL------SRQRRQFVFP---- 34
SNAK_DROME 49 EQPVATIGEDVDDQDTEDEESYLKFGDDAEVRTSVSEGLHEGAFCRRSFD 98
|||. .|...|.
PCE_TACTR 35 -----------------DEEE----------------------LCSNRFT 45
SNAK_DROME 99 GRSGYCILAYQCLHVIREYRVHGTRIDICTHRNNVPVICCPLADKHVLAQ 148
..|.|.....|..::::...:..:..||......|.:||| ...||
PCE_TACTR 46 -EEGTCKNVLDCRILLQKNDYNLLKESICGFEGITPKVCCP-KSSHV--- 90
SNAK_DROME 149 RISATKCQEYNAAARRLHLTDTGRTFSGKQCVPSVP-----------LIV 187
||:|:.........| ..||..|::| .|:
PCE_TACTR 91 -ISSTQAPPETTTTER----------PPKQIPPNLPEVCGIHNTTTTRII 129
SNAK_DROME 188 GGTPTRHGLFPHMAALGWTQGSGSKDQDIKWGCGGALVSELYVLTAAHCA 237
||.....|.:|.|.|:...||.....| ||||||:..:|:||:||.
PCE_TACTR 130 GGREAPIGAWPWMTAVYIKQGGIRSVQ-----CGGALVTNRHVITASHCV 174
SNAK_DROME 238 TSGS----KPPDM--VRLGARQLNET--SATQQDIKILIIVLHPKYRSSA 279
.:.: .|.|: ||||...|..| .:...|..:..:..|..:..:.
PCE_TACTR 175 VNSAGTDVMPADVFSVRLGEHNLYSTDDDSNPIDFAVTSVKHHEHFVLAT 224
SNAK_DROME 280 YYHDIALLKLTRRVKFSEQVRPACL----WQLPELQIPTVVAAGWGRTEF 325
|.:|||:|.|...|.|::::||.|| .:..:|.:......|||.|.|
PCE_TACTR 225 YLNDIAILTLNDTVTFTDRIRPICLPYRKLRYDDLAMRKPFITGWGTTAF 274
SNAK_DROME 326 LGAKSNALRQVDLDVVPQMTCKQIYRKERRLPRGIIEGQFCAGYLPGGRD 375
.|..|..||:|.|.:.....|:|.|.|: ..|.....|||:..||:|
PCE_TACTR 275 NGPSSAVLREVQLPIWEHEACRQAYEKD----LNITNVYMCAGFADGGKD 320
SNAK_DROME 376 TCQGDSGGPIHALLPEYNCVAFVVGITSFGKFCAAPNAPGVYTRLYSYLD 425
.||||||||: :||......:::||.||||.||.|..|||||::..:||
PCE_TACTR 321 ACQGDSGGPM--MLPVKTGEFYLIGIVSFGKKCALPGFPGVYTKVTEFLD 368
SNAK_DROME 426 WIEKIAFKQH 435
|| .:|
PCE_TACTR 369 WI-----AEHMV 375
Hay que utilizar un alineamiento global sólo cuando sabemos que las dos secuencias se parecen a lo largo de toda su extensión. Esto lo podemos averiguar con un dot plot.
Más tarde fue adaptado por Smith y Waterman para alineamientos locales.
Los alineamientos locales se utilizan cuando sólo tenemos un fragmento de la secuencia o cuando las secuencias son distantes y sólo una parte se parece.
########################################
# Program: water
# Rundate: Tue Oct 02 11:00:39 2007
# Align_format: srspair
# Report_file: /ebi/extserv/old-work/water-20071002-11003873398600.output
########################################
#=======================================
#
# Aligned_sequences: 2
# 1: SNAK_DROME
# 2: PCE_TACTR
# Matrix: EBLOSUM62
# Gap_penalty: 10.0
# Extend_penalty: 0.5
#
# Length: 362
# Identity: 116/362 (32.0%)
# Similarity: 165/362 (45.6%)
# Gaps: 50/362 (13.8%)
# Score: 452.0
#
#
#=======================================
SNAK_DROME 89 EGAFCRRSFDGRSGYCILAYQCLHVIREYRVHGTRIDICTHRNNVPVICC 138
|...|...|. ..|.|.....|..::::...:..:..||......|.:||
PCE_TACTR 36 EEELCSNRFT-EEGTCKNVLDCRILLQKNDYNLLKESICGFEGITPKVCC 84
SNAK_DROME 139 PLADKHVLAQRISATKCQEYNAAARRLHLTDTGRTFSGKQCVPSVP---- 184
| ...|| ||:|:.........| ..||..|::|
PCE_TACTR 85 P-KSSHV----ISSTQAPPETTTTER----------PPKQIPPNLPEVCG 119
SNAK_DROME 185 -------LIVGGTPTRHGLFPHMAALGWTQGSGSKDQDIKWGCGGALVSE 227
.|:||.....|.:|.|.|:...||.....| ||||||:.
PCE_TACTR 120 IHNTTTTRIIGGREAPIGAWPWMTAVYIKQGGIRSVQ-----CGGALVTN 164
SNAK_DROME 228 LYVLTAAHCATSGS----KPPDM--VRLGARQLNET--SATQQDIKILII 269
.:|:||:||..:.: .|.|: ||||...|..| .:...|..:..:
PCE_TACTR 165 RHVITASHCVVNSAGTDVMPADVFSVRLGEHNLYSTDDDSNPIDFAVTSV 214
SNAK_DROME 270 VLHPKYRSSAYYHDIALLKLTRRVKFSEQVRPACL----WQLPELQIPTV 315
..|..:..:.|.:|||:|.|...|.|::::||.|| .:..:|.:...
PCE_TACTR 215 KHHEHFVLATYLNDIAILTLNDTVTFTDRIRPICLPYRKLRYDDLAMRKP 264
SNAK_DROME 316 VAAGWGRTEFLGAKSNALRQVDLDVVPQMTCKQIYRKERRLPRGIIEGQF 365
...|||.|.|.|..|..||:|.|.:.....|:|.|.|: ..|.....
PCE_TACTR 265 FITGWGTTAFNGPSSAVLREVQLPIWEHEACRQAYEKD----LNITNVYM 310
SNAK_DROME 366 CAGYLPGGRDTCQGDSGGPIHALLPEYNCVAFVVGITSFGKFCAAPNAPG 415
|||:..||:|.||||||||: :||......:::||.||||.||.|..||
PCE_TACTR 311 CAGFADGGKDACQGDSGGPM--MLPVKTGEFYLIGIVSFGKKCALPGFPG 358
SNAK_DROME 416 VYTRLYSYLDWI 427
|||::..:||||
PCE_TACTR 359 VYTKVTEFLDWI 370
Los alineamientos globales se utilizan sobre todo dentro de alineamientos múltiples.
Los alineamientos locales generan alineamientos de las zonas más similares de las secuencias.
El algoritmo genera una matriz que representa todos los posibles alineamientos entre las dos secuencias, de modo análogo al gráfico del dot matrix, pero ahora en vez de dibujar puntos se asignan puntuaciones.
Se comienza en los extremos de las secuencias y se intenta alinear todos los posibles pares de caracteres en las secuencias dando una puntuación a las coincidencias, las no coincidencias y los gaps.
El mejor conjunto de puntuaciones altas consecutivas en la matriz define el alineamiento óptimo.
El alineamiento dependerá de los parámetros utilizados: gaps, coincidencias, no coincidencias y similaridad.
Es un método matemáticamente muy estricto, pero lento.
El tiempo de ejecución crece con el producto de las longitudes de las secuencias a alinear.
Hay muchos programas que utilizan estos algoritmos. Dos de los más utilizados son needle y water del EMBOSS.
Matrices de sustitución¶
Dos nucleótidos pueden o ser iguales o diferentes, pero en el caso de los aminoácidos la situación es más compleja.
Dos aminoácidos pueden ser iguales, diferentes o más o menos parecidos. Por ejemplo un aminoácido neutro se parecerá más a otro aminoácido neutro que a uno ácido.
Ejemplo de posible sistema de puntuación para aminoácidos.
* Tipos de aminoácidos:
hidrofóbicos: Ile, Val, Leu, Ala
Polares (+): Lys, Arg
Polares (-): Glu, Asp
Aromáticos: Phe, Tyr, Trp
etc.
* Puntuaciones:
Ile x Val = -1
Ile x Asp = -5
Phe x Tyr = -1
Phe x Gly = -8
Una forma de cuantificar el parecido entre diferentes aminoácidos es estudiar cuantas veces suelen aparecer en la misma posición en proteínas homólogas.
Si los dos aminoácidos suelen aparecer en las mismas posiciones debe ser porque funcionalmente son más equivalentes que dos aminoácidos que no suelen aparecer en las mismas posiciones.
En estas matrices se comparan todos los aminoácidos con todos los demás. A cada comparación se le da una puntuación que indica como es de frecuente que un aminoácido sea sustituido por otro en proteínas homólogas.
Una de estas matrices es la PAM (Percent Accepted Mutation).
Hay matrices PAM preparadas a partir de comparaciones entre secuencias que se parecen mucho y entre secuencias que se parecen menos.
PAM 250 se utiliza para secuencias proteicas que se parecen muy poco entre sí (~25%), para secuencias que se parecen un 40%, 50% o 60% es mejor utilizar PAM120, PAM80 Y PAM60 respectivamente.
Otras matrices utilizadas para comparar proteínas son las BLOSUM.
Están preparadas a partir de alineamientos de regiones conservadas de proteínas llamadas bloques.
Las hay preparadas a partir de bloques con un 80% o 60% de identidad, BLOSUM80 y BLOSUM 60 respectivamente.
Las PAM se suelen utilizar en estudios filogenéticos y las BLOSUM en búsquedas de dominios conservados.
Existen otras matrices de sustitución creadas con fines específicos, por ejemplo para buscar proteínas con estructuras similares.
Penalizaciones por gaps (Gap penalties)¶
Sirven para indicar al algoritmo si queremos favorecer la aparición de gaps en el alineamiento o no.
Normalmente se pueden modificar dos gap penalties, el de creación de un nuevo gap y el de extensión del gap.
Si se ponen unos gap penalties muy altos no aparecerán gaps en el alineamiento, si son muy bajos aparecerán demasiados.
Es una buena idea utilizar los valores por defecto a no ser que tengamos alguna razón para cambiarlos.
Significación de los alineamientos¶
Siempre que alimentemos una algoritmo de alineamiento con un par de secuencias obtendremos un alineamiento, incluso aunque las secuencias estén compuestas por letras al azar y no se parezcan nada entre sí.
Por lo tanto además de hacer el alineamiento hay que estimar la significación estadística del mismo.
Si trabajamos con secuencias de ADN es fácil hacerse una idea de lo bueno que es el alineamiento puesto que al valorar positivamente sólo la identidad los alineamientos obtenidos por azar suelen ser muy cortos.
Con las secuencias de proteínas el asunto es más complejo porque se valoran positivamente también los parecidos de aminoácidos que no son idénticos, pero que son químicamente similares y esto conduce a que los alineamientos de las secuencias al azar puedan confundirse con alineamientos realmente significativos.
Casi todos los programas de alineamiento suelen calcular también índices que indican la significación. Hay que estudiar estos índices para no confundir alineamientos espúreos con reales.

{kind=link}
{kind=link}