Búsqueda de secuencias utilizando BLAST¶
La familia de programas BLAST es la más utilizada para buscar secuencias similares en una base de datos dada una secuencia problema.
Esta práctica consta de dos partes, en la primera nos familiarizaremos con el interfaz web del servidor BLAST del NCBI y en la segunda utilizaremos BLAST para resolver algunos problemas prácticos.
BLAST en el NCBI¶
Formulario de búsqueda¶
La página principal del BLAST en el NCBI nos permite elegir directamente distintos organismos (Human, Mouse, etc.), distintos programas (blastn, blastp, etc.) y otras búsquedas más especializadas.
Vamos a hacer un blast con ADN (nucleotide blast) utilizando una secuencia de ejemplo de rata.
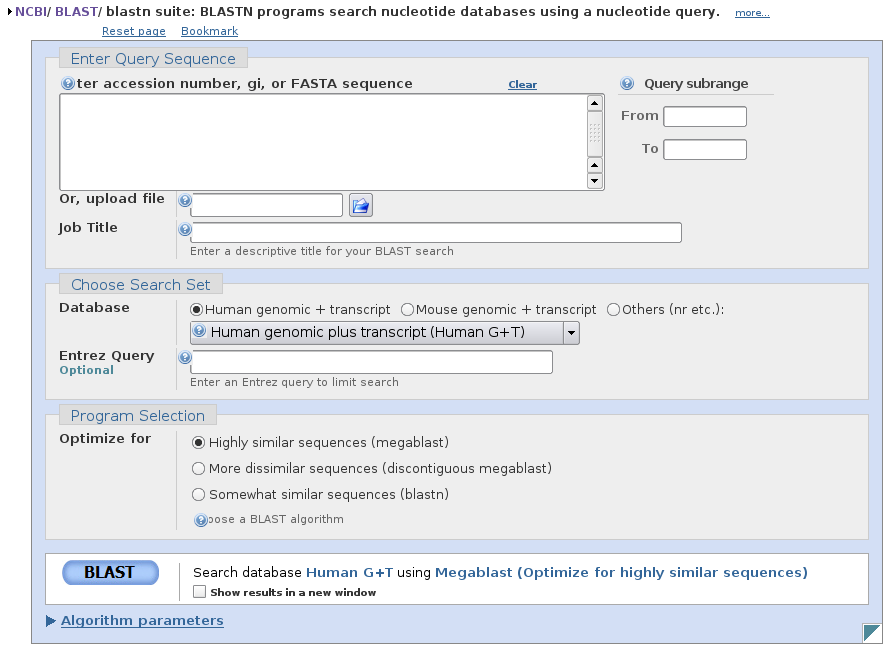
En la página de búsqueda del BLAST podemos modificar numerosos parámetros.
El más importante de ellos es la secuencia que queremos utilizar en la búsqueda (Enter Query Sequence). Podemos poner una secuencia en formato fasta o un número de acceso de la Genbank. Además podemos limitar la búsqueda a una región concreta de la secuencia (Query subrange). El formulario nos permite también escoger un fichero que contenga la secuencia.
La segunda decisión importante es la base de datos con la que vamos a comparar nuestra secuencia (Choose Search Set). Podemos elegir una de las numerosas bases de datos ofrecidas por el NBCI (humano, ratón, nr, refseq, etc.) o podemos escribir una expresión de búsqueda para el entrez. Si elegimos esta última opción la búsqueda se realizará en comparando nuestra secuencia con las secuencias resultantes de esta búsqueda.
Por último, podemos seleccionar el programa a utilizar: megablast (para encontrar secuencias muy similares), discontiguous megablast (para secuencias algo diferentes) y blast para secuencias algo más distintas. Cuanto más sensible sea el algoritmo más tiempo tardará la búsqueda.
Como ejemplo realizar una búsqueda con megablast sobre la base de datos humana.
Mientras el BLAST se está ejecutando veremos una página en la que se nos informa sobre el tiempo estimado que requerirá la búsqueda.
Página de resultados¶
El resultado del BLAST se divide en varias secciones. En la primera se nos informa sobre la versión del programa utilizado, el nombre de la base de datos y el número de secuencias buscadas.

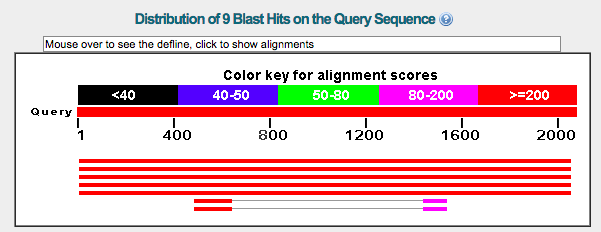
A continuación aparecen una figura y una tabla con un resumen de los resultados. En la figura se observa una fila para cada una de las secuencias similares obtenidas (hit). En cada fila se representa la región cubierta por los distintos HSPs. Estas regiones están pintadas de un color u otro dependiendo del E-value correspondiente.
¿Cuántas secuencias similares se han obtenido?
¿Cuantos HSPs tiene el primer hit, y el segundo y el tercero?
¿Tienen todos los HSPs el mismo E-value?
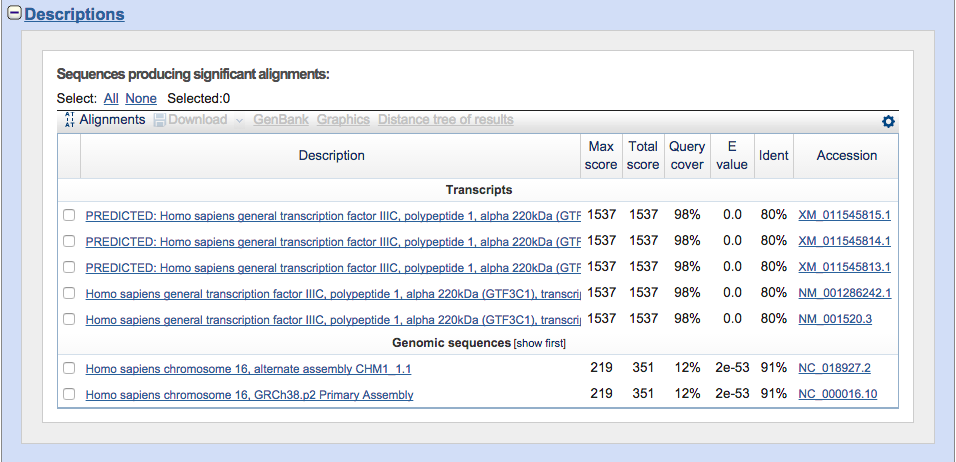
En la tabla también se encuentran los distintos resultados (hits). Para cada uno se puede observar la definición de la secuencia (description) y el E-value. En la tabla los hits están ordenados por el E-value, de menor a mayor.
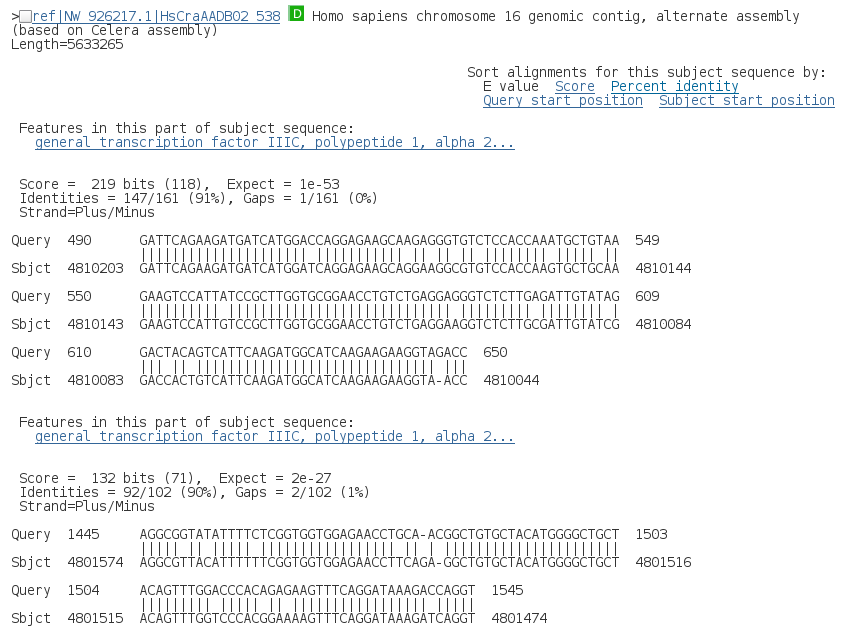
En la siguiente sección de la página de resultados se muestran los alineamientos de la secuencia problema (query) con las distintas secuencias de la base de datos (Sbjct). Para cada una de ellas se muestra la descripción y los alineamientos correspondientes a los distintos HSPs.
¿Cuantos alineamientos distintos hay para cada secuencia?
¿En el caso de haber distintos HSPs tienen todos el mismo E-value?
¿Con que criterio están ordenados los HSPs dentro de cada secuencia?
¿Qué E-value aparece en la tabla resumen?
Anotación de secuencias¶
Hemos obtenido en un proyecto de secuenciación de Amblyomma variegatum secuencias de varios genes (1, 2, 3, 4) y queremos saber cual es la función de cada una de ellas.
¿Qué tipo de base de datos sería mejor utilizar ADN o proteína?
¿Qué programa habría que utilizar?
Una buena base de datos para este menester es la refseq, una base de datos de referencia para proteínas.
Muchas de las proteínas similares están marcadas como predicted o hypothetical protein.
¿Qué puede significar esto? ¿Pasaría lo mismo si hubiésemos utilizado la swissprot? ¿Habría sido mejor utilizar la swissprot?
Clasificación¶
Nos envían de un hospital una muestra de sangre de un paciente para que ayudemos en el diagnóstico. Al parecer tiene una enfermedad causada por un microorganismo, ¿por cual?.
Para poder solucionar el problema extraemos ADN genómico a partir de la sangre del paciente y hacemos una PCR utilizando unos cebadores diseñados para amplificar la región ITS1.
Esta región ha sido ampliamente utilizada para clasificar todo tipo de organismos y se dispone de numerosas secuencias en la base de datos.
Una vez obtenida la PCR la enviamos a secuenciar y obtenemos la secuencia para un fragmento del ITS1.
¿Qué programa convendría utilizar en la búsqueda y qué base de datos?
¿De qué especie es la infección? ¿Es seria la enfermedad? ¿Se puede precisar la cepa (strain) con esta secuencia?
Teniendo en cuenta que la tasa de error en la secuenciación se encuentra alrededor del 2%, ¿conviene gastar el dinero en amplificar y secuenciar otras secuencias para asegurarse de la especie del microorganismo o es suficiente con lo que hemos obtenido?
Filogenia¶
Queremos establecer la filogenia dentro de los animales y para ello decidimos utilizar la secuencia del ARN mensajero de la frataxina.
¿Qué ventajas y desventajas tendría utilizar la base de datos nr o la refseq mRNA?
Utilizando la refseq, ¿Se obtiene el mismo resultado utilizando blastn y tblastx? ¿Cuántas secuencias se obtienen en uno y otro caso? ¿Por qué?
En el resultado con blastn con la base de datos refseq sólo se obtienen secuencias de primates y en el de tblastx se obtiene incluso la frataxina de Xenopus.
En el resultado obtenido con tblastx la frataxina de Xenopus tiene un E-value de 2e-56, ¿por qué hay secuencias posteriores de primates que presentan un E-value mayor?
Una vez hemos hecho el BLAST podemos obtener todas las secuencias del resultado haciendo click en “Select All” y en “Get selected sequences”.
