Búsqueda de secuencias en bases de datos¶
Una vez que sabemos alinear dos secuencias podemos alinear una secuencia con todas las de una base de datos.
Si buscamos los alineamientos significativos encontramos qué secuencias se parecen a nuestra secuencia query.
Para hacer esto se necesita un algoritmo muy rápido porque hay millones de alineamientos que procesar.
BLAST (Basic Local Alignment Search Tool) utiliza un algoritmo muy rápido basado en palabras (word, k-tuple o kmers)
BLAST¶
BLAST se utiliza habitualmente para comparar una secuencia dada contra toda una base de datos de millones de secuencias.
Producen alineamientos locales entre cada pareja de secuencias.
Además generan un valor se significación estadística para cada alineamiento.
Algoritmo¶
Si se utilizase un algoritmo como el de Smith y Waterman para alinear un secuencia de N letras con una base de datos de M letras se necesitarían NxM comparaciones.
Para simplificar la búsqueda, antes de comenzar, se genera un índice de todas las palabras cortas contenidas en la secuencia problema.
DSFASPUIVH
DSF
SFA
FAS
ASP
SPU
PUI
UIV
IVH
Se buscan las secuencias de la base de datos que presentan alguna de las palabras de la lista anterior.
El tamaño de las palabras influye en la sensibilidad del BLAST.
Cuanto más pequeñas sean las palabras más sensible será la búsqueda, pero más lenta.
Se buscan diagonales en la matriz que presenten palabras comunes. A estos fragmentos se les denomina high scoring pair (HSP).
Los HSP se extienden hasta que la puntuación del alineamiento comienza a disminuir.
Si el HSP acaba teniendo un valor de significación mayor que el dado se incluirá en el alineamiento final.
Con este algoritmo la búsqueda requiere (N-2) + (M -2) operaciones en vez de NxM.
Para determinar si un alineamiento es significativo se compara su puntuación con la de alineamientos de secuencias al azar.
e-value¶
Una vez obtenido el alineamiento hay que preguntarse si es significativo.
Alineamiento obtenido entre dos proteínas seleccionadas al azar.
Query= unknown
(207 letters)
>unknown
Length = 333
Score = 14.2 bits (25), Expect = 2.9
Identities = 4/16 (25%), Positives = 8/16 (50%)
Query: 160 PKRYDWTGKNWVYSHD 175
P ++ W +SH+
Sbjct: 146 PTKHAWQEIGKEFSHE 161
¿Cual es la probabilidad de que un alineamiento con una puntuación (score) similar se obtenga por azar entre dos secuencias no relacionadas.
El e-value (Expect) es el número de alineamientos que esperamos para una puntuación (score) X (o superior) en la búsqueda que estamos realizando si la base de datos fuese una colección de letras al azar.
Para calcular esta probabilidad se pueden generar secuencias al azar de la misma longitud y composición que la query y se alinean.
Si la base de datos es suficientemente grande y contiene mayoritariamente secuencias no relacionadas la distribución de scores observados debe coincidir con la distribución de scores esperados por azar.
A partir de estos alineamientos se estudia la distribución de scores.
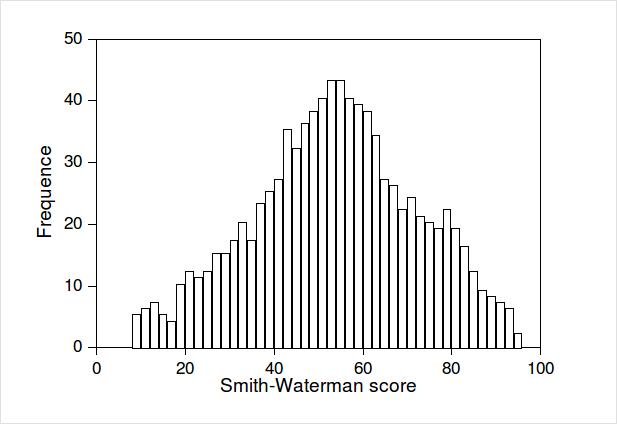
Se espera encontrar muchos alineamientos al azar con puntuaciones bajas, pero muy pocos con puntuaciones altas.
Para puntuaciones muy altas se espera encontrar un número insignificante de alineamientos por simple azar y los e-values son, por lo tanto, muy bajos.
El valor e-value dado por el blast depende de la base de datos empleada y de la longitud de la secuencia.
El número de alineamientos con un score > S se incrementa linealmente con el tamaño de la base de datos.
Una secuencia, un alineamiento con un score S, encontrada en una búsqueda contra un genoma bacteriano con 1000-5000 secuencias va a ser 50-250 veces más significativa que un alineamiento con exactamente el mismo score en una base de datos con 250,000.
En búsquedas reales son comunes e-values de entre 10e-6 y 10e-100.
Ejemplo de BLAST¶
Blast of sequence c15d_01-D10-M13R_c against database arabidopsis
BLASTX 2.2.10 [Oct-19-2004]
Reference: Altschul, Stephen F., Thomas L. Madden, Alejandro A. Schaffer,
Jinghui Zhang, Zheng Zhang, Webb Miller, and David J. Lipman (1997),
"Gapped BLAST and PSI-BLAST: a new generation of protein database search
programs", Nucleic Acids Res. 25:3389-3402.
Query= c15d_01-D10-M13R_c
(892 letters)
Database: tair6
30,690 sequences; 12,653,157 total letters
Searching..................................................done
Score E
Sequences producing significant alignments: (bits) Value
AT3G28480.1 | Symbol: None | oxidoreductase, 2OG-Fe(II) oxygenas... 280 6e-76
AT3G28490.1 | Symbol: None | oxidoreductase, 2OG-Fe(II) oxygenas... 258 3e-69
AT5G18900.1 | Symbol: None | oxidoreductase, 2OG-Fe(II) oxygenas... 244 4e-65
AT3G06300.1 | Symbol: AT-P4H-2 | Encodes a prolyl-4 hydroxylase ... 234 4e-62
AT1G20270.1 | Symbol: None | oxidoreductase, 2OG-Fe(II) oxygenas... 213 1e-55
AT4G35810.1 | Symbol: None | oxidoreductase, 2OG-Fe(II) oxygenas... 201 4e-52
AT2G17720.1 | Symbol: None | oxidoreductase, 2OG-Fe(II) oxygenas... 192 2e-49
AT5G66060.1 | Symbol: None | oxidoreductase, 2OG-Fe(II) oxygenas... 186 2e-47
AT4G35820.1 | Symbol: None | oxidoreductase, 2OG-Fe(II) oxygenas... 159 2e-39
AT4G33910.1 | Symbol: None | oxidoreductase, 2OG-Fe(II) oxygenas... 149 3e-36
AT2G43080.1 | Symbol: AT-P4H-1 | Encodes a prolyl-4 hydroxylase ... 147 1e-35
AT2G23096.1 | Symbol: None | oxidoreductase, 2OG-Fe(II) oxygenas... 137 7e-33
AT4G25600.1 | Symbol: None | ShTK domain-containing protein, sim... 131 6e-31
>AT3G28480.1 | Symbol: None | oxidoreductase, 2OG-Fe(II) oxygenase family
protein, similar to prolyl 4-hydroxylase, alpha subunit,
from Gallus gallus (GI:212530), Rattus norvegicus
(GI:474940), Mus musculus (SP:Q60715); contains PF03171
2OG-Fe(II) oxygenase superfamily domain |
chr3:10677275-10679525 REVERSE | Aliases: MFJ20.16
Length = 316
Score = 280 bits (717), Expect = 6e-76
Identities = 141/235 (60%), Positives = 174/235 (74%), Gaps = 1/235 (0%)
Frame = +2
Query: 191 NRFPKMLLHNNDMYESVIRMKTGGSAITIDPTRVIQLSSKPRAFLYEGFLSYEECQHLIH 370
NRF + +N SVI+MKT S+ DPTRV QLS PR FLYEGFLS EEC H I
Sbjct: 25 NRF--LTRSSNTRDGSVIKMKTSASSFGFDPTRVTQLSWTPRVFLYEGFLSDEECDHFIK 82
Query: 371 LAKGKLRQSLVAAG-TGESVASKERTSTGMFLRKAQGKIVARIESRIAAWTFLPLDNGEP 547
LAKGKL +S+VA +GESV S+ RTS+GMFL K Q IV+ +E+++AAWTFLP +NGE
Sbjct: 83 LAKGKLEKSMVADNDSGESVESEVRTSSGMFLSKRQDDIVSNVEAKLAAWTFLPEENGES 142
Query: 548 IQILRYENGQKYEPHFDFFQDPGNIAIGGHRIATILMYLSDVEKGGETVFPNSPVKLSEE 727
+QIL YENGQKYEPHFD+F D N+ +GGHRIAT+LMYLS+VEKGGETVFP K ++
Sbjct: 143 MQILHYENGQKYEPHFDYFHDQANLELGGHRIATVLMYLSNVEKGGETVFPMWKGKATQL 202
Query: 728 EKGDLSECAXVGYGVRPKLGDALLFFSMNPNVTPDATSYHGSCPVIEGEKMVCTK 892
+ +ECA GY V+P+ GDALLFF+++PN T D+ S HGSCPV+EGEK T+
Sbjct: 203 KDDSWTECAKQGYAVKPRKGDALLFFNLHPNATTDSNSLHGSCPVVEGEKWSATR 257
>AT3G28490.1 | Symbol: None | oxidoreductase, 2OG-Fe(II) oxygenase family
protein, similar to prolyl 4-hydroxylase, alpha subunit,
from Caenorhabditis elegans (GI:607947), Mus musculus
(SP:Q60715), Homo sapiens (GI:18073925); contains
PF03171 2OG-Fe(II) oxygenase superfamily domain |
chr3:10680286-10681891 REVERSE | Aliases: MFJ20.17
Length = 288
Score = 258 bits (659), Expect = 3e-69
Identities = 128/211 (60%), Positives = 159/211 (75%), Gaps = 2/211 (0%)
Frame = +2
Query: 266 AITIDPTRVIQLSSKPRAFLYEGFLSYEECQHLIHLAKGKLRQSLVAAG--TGESVASKE 439
+ ++DPTR+ QLS PRAFLY+GFLS EEC HLI LAKGKL +S+V A +GES S+
Sbjct: 24 SFSVDPTRITQLSWTPRAFLYKGFLSDEECDHLIKLAKGKLEKSMVVADVDSGESEDSEV 83
Query: 440 RTSTGMFLRKAQGKIVARIESRIAAWTFLPLDNGEPIQILRYENGQKYEPHFDFFQDPGN 619
RTS+GMFL K Q IVA +E+++AAWTFLP +NGE +QIL YENGQKY+PHFD+F D
Sbjct: 84 RTSSGMFLTKRQDDIVANVEAKLAAWTFLPEENGEALQILHYENGQKYDPHFDYFYDKKA 143
Query: 620 IAIGGHRIATILMYLSDVEKGGETVFPNSPVKLSEEEKGDLSECAXVGYGVRPKLGDALL 799
+ +GGHRIAT+LMYLS+V KGGETVFPN K + + S+CA GY V+P+ GDALL
Sbjct: 144 LELGGHRIATVLMYLSNVTKGGETVFPNWKGKTPQLKDDSWSKCAKQGYAVKPRKGDALL 203
Query: 800 FFSMNPNVTPDATSYHGSCPVIEGEKMVCTK 892
FF+++ N T D S HGSCPVIEGEK T+
Sbjct: 204 FFNLHLNGTTDPNSLHGSCPVIEGEKWSATR 234
Versiones del BLAST¶
Existen varios programas BLAST dependiendo de las secuencias que queramos comparar:
BLASTP: compara proteínas con una base de datos de proteínas.
BLASTN: compara nucleótidos con una base de datos de nucleótidos.
BLASTX: compara nucleótidos (antes los traduce) con una base de datos de proteínas.
TBLASTN: compara proteínas contra una base de datos de nucleótidos (antes los traduce).
TBLASTX: compara nucleótidos con una base de datos de nucleótidos traduciendo todas las secuencias.
BLASTN se utiliza para comparar secuencias de nucleótidos próximas filogenéticamente. BLASTN no se recomienda para buscar proteínas homólogas de otros organismos. Es mejor hacer las búsquedas con proteínas.
Cuando las secuencias son muy distintas y codifican proteínas TBLASTX puede dar mejores resultados, aunque es más costoso computacionalmente. Las búsquedas con proteínas son más sensibles y permiten encontrar secuencias más alejadas evolutivamente. Al estar el código genetico degenerado los cambios en el ADN no siempre se traducen en un cambio en las proteínas.
Las versiones de BLAST que realizan traducciones lo hacen en las 6 pautas de lectura posibles.
Megablast¶
Utiliza un algoritmo modificado para encontrar alineamientos largos entre secuencias muy similares.
Es mucho más rápido que BLAST, pero algo menos sensible.
Se utiliza para encontrar secuencias muy similares a la secuencia problema.
Si queremos comprobar si una secuencia ya está descrita en la base de datos esta es la mejor opción.
BLAST 2 Sequences¶
Utiliza el algoritmo del blast para alinear dos secuencias.
No utiliza ninguna base de datos.
PSI-BLAST¶
Position-Specific Iterated (PSI)-BLAST es el BLAST más sensible.
Es capaz de encontrar proteínas muy alejadas filogenéticamente.
PSI-BLAST parte de un BLAST de proteína con proteína estándar.
A partir del alineamiento de los mejores resultados construye una matrix PSSM (Position-specific scoring matrix) (también conocido como Profile o Motif (motivo)).
En una matriz de puntuación PSSM las puntuaciones de cambio entre aminoácidos dependen de la posición en el alineamiento.
Un cambio Tyr-Trp puede tener una puntuación diferente en dos posiciones distintas de la secuencia.
Los PSSM o profiles se utilizan para describir dominios conservados en las proteínas.
Para cada posición se almacena cuáles son las frecuencias de cada carácter.
Ejemplo de dominio conservado.
1B7B_B 3 .[1].KMVVALGGNAILS.[11].LVQTSA.[ 7].Q.[1].HRLIVSHGNG.[1].Q.[1].GNLLLQQQAAD.[ 3]. 69
gi 22653947 1 .[1].RIVITIGGSIIIS.[ 6].FRAYAD.[ 7].E HDLFVVVGGG.[1].P ARDYIGVAREL.[ 1]. 58
gi 81344717 1 .[1].VTVLSLGGSIVAP.[ 7].LGRFVR.[ 9].A.[1].RKLIVVSGGG.[1].P ARTYQNAYRAL.[27]. 88
gi 81342918 1 .[1].LEIISLGGGVINS.[ 7].IKNFKN.[ 9].E.[1].RKIILIVGGG.[1].V AREYQDAYKKI.[ 3]. 64
gi 2495997 1 MHIVKIGGSLTYD.[ 3].LLKALK.[ 5].N.[1].KKIVIIPGGG.[1].F ANVVRKIDKAL.[ 1]. 53
gi 74510802 13 .[1].EWVVKIGGSLFPE.[ 3].ALLESL E.[1].TGAAVVCGGG.[1].F ADMVRYHDRKF.[ 1]. 61
gi 2495760 1 .[1].LTILKLGGSILSD.[11].LERIAM.[13].K.[1].IKLILVHGGG A.[1].GHPVAKKYLKI.[12]. 81
gi 74507521 1 MIILKLGGSVITR.[12].LERIAS.[ 6].P SSLMIVHGAG S.[1].GHPFAGEYRIG.[14]. 75
gi 74571552 1 MIIIKIGGSVLSD.[11].VKSIAY.[ 6].P.[1].EKFIVVHGGG S.[1].GHPLAKEFAIR.[10]. 71
gi 74548980 7 .[1].VTILKIGGSVITD.[ 9].KERELR.[ 8].K.[1].RNLIVVHGAG S.[1].GHPHVKKFGLS.[ 2]. 70
En la segunda iteración el PSSM se convierte en la query de la búsqueda.
Las secuencias encontradas en la nueva búsqueda se incluyen en un nuevo PSSM que se utiliza de nuevo para hacer otra búsqueda.
La búsqueda PSI-BLAST concluye cuando ya no se encuentran nuevas secuencias en las posteriores iteraciones.
