Ensamblaje y mapeo de secuencias tipo Sanger¶
Hasta ahora en el curso hemos hablado de como analizar secuencias de genes, genomas, de mensajeros. Pero no hemos tenido en cuenta como se obtienen esas secuencias. Todas las tecnologías de secuenciación producen fragmentos más o menos cortos y a partir de estos se debe recomponer la secuencia del fragmento analizado, ya sea un genoma, un BAC o un producto de PCR. Este proceso se denomina proyecto de secuenciación. En muchos de los casos el resultado no será la secuencia completa del fragmento y será necesario completar los huecos entre los diferentes conjuntos de secuencias obtenidos.
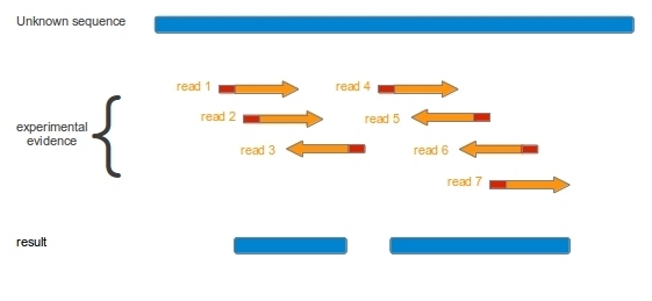
Ensamblaje de secuencias¶
El proceso de ensamblaje consta de tres fases: limpieza de secuencias, alineamiento de secuencias y ensamblaje.
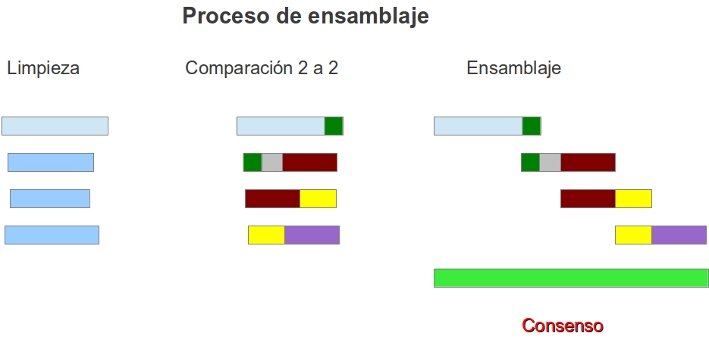
Limpieza¶
Es el primer paso a realizar ante cualquier proyecto de secuenciación. Los secuenciadores devuelven las secuencias crudas y suele ser necesario eliminar o bloquear diferentes regiones que contengan:
bases con mala calidad de lectura
bases que pertenezcan a vectores o adaptadores
bases que estén altamente repetidas (secuencias simples o elementos transponibles)
contaminantes
Estas regiones interfieren con el ensamblaje y en la mayoría de los programas de ensamblaje es necesario eliminarlas o enmascarlas antes de comenzar.
Alineamiento de secuencias¶
El siguiente paso es alinear todas las secuencias con todas las secuencias dos a dos para identificar que secuencias solapan entre si. Vamos a identificar pares o grupos de secuencias con fragmentos de secuencias comunes. El resultado es una red de interacciones entre las secuencias que alinean entre si, en el que en cada borde está localizado una secuencia.
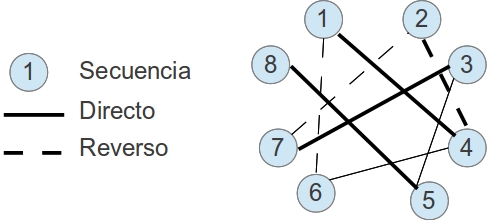
Ensamblaje de secuencias¶
A partir de estos grafos realizados con las secuencias identificadas que comparten regiones comunes, se crea el ensamblaje. A diferencia de un alineamiento múltiple los ensambladores no fuerzan que todas las secuencias cubran la totalidad del alineamiento, este se va alargando con la adición de nuevas secuencias. Existen diferentes algoritmos que a partir de la información almacenada en el grafo ordenan y ensamblar las secuencias utilizando alineación múltiple para obtener la mejor distribución de las secuencias solapantes. En muchos programas estos dos últimos pasos (alineamientos de secuencias y ensamblaje) son cíclicos. A partir de estos alineamientos se obtienen la secuencia consenso que representa a cada conjunto de secuencias o contig. Existen diferentes reglas para construir el consenso, pero es indispensable que tengan en cuenta el valor de la calidad de las secuencias.
Problemas del ensamblaje de secuencias¶
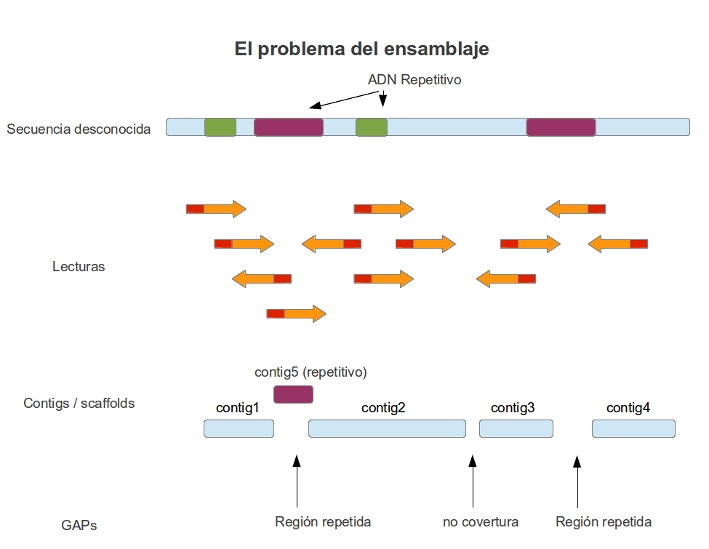
Hay varios factores que afectan a la calidad del ensamblaje, la calidad de las secuencias, el número de veces que se lee cada posición (cobertura), la longitud de las secuencias y la existencia de secuencias repetidas.
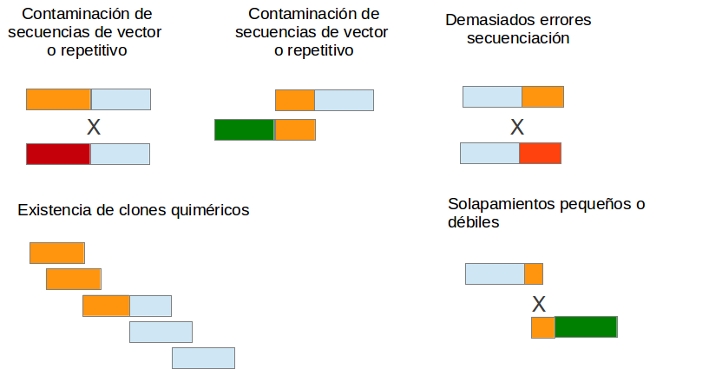
Para obtener buenos ensamblajes hay que obtener buenos solapamientos (overlaps) entre nuestras lecturas. Si dos lecturas solapan suponemos que provienen de la misma región secuenciada, cuanto mayor y con mayor similitud sea el solapamiento mejor. Pero se pueden producir solapamientos debido al azar, regiones repetidas o genes duplicados. Para evitar estos falsos positivos podemos tener en cuenta una serie de factores:
Similitud de la región solapada.
Longitud de la región solapada
Porcentaje de secuencia que solapa
Ignorar secuencias que solapan con muchas secuencias (repetitivas)
Pero hay que ajustar estos parámetros ya que valores alto de los threshold podrían hacer perder solapamientos, sobretodo en regiones con baja cobertura o si la muestra contiene mucha variabilidad (secuenciación de pools de individuos, metagenómica, etc)
Los errores causados por la mala calidad, escasa cobertura, longitud de secuencias son evitables mediante la limpieza de secuencias o la adición de nuevas secuencias al ensamblaje. Pero el problema de las secuencias repetidas no puede obviarse ya que en los genomas, sobretodo en los eucariotas, existe un alto porcentaje de secuencias repetidas.
Secuencias repetidas¶
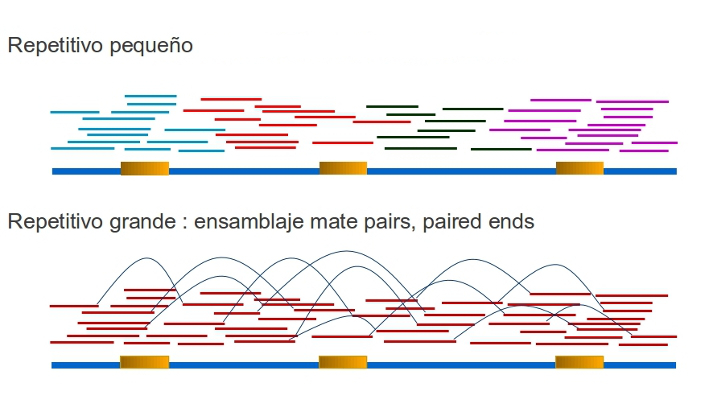
Las lecturas que únicamente contienen el mismo ADN repetitivo ensamblaran entre si aunque provengan de regiones distintas del genoma, solo podremos diferenciarlas si contienen alguna parte de secuencia única. Esto hace que si la longitud del ADN repetitivo es mayor que el tamaño de las lecturas los ensambladores no puedan ensamblar estas regiones ya que solapan con multitud de copias, esto crea gaps y diferentes contigs. Las regiones repetidas más pequeñas que la longitud de las secuencias se podrán ensamblar perfectamente siempre que tengamos una lectura que la cruce en su totalidad, es decir que tenga en sus extremos secuencias de copia única. Para obtenerlas es necesario una correcta cobertura.
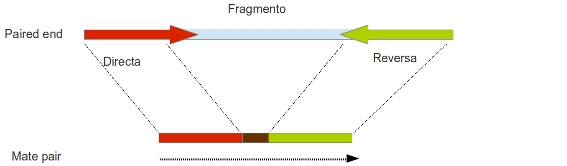
Para solventar el problema de la repeticiones de mayor tamaño que las lecturas, es necesario recurrir a distintas estrategias experimentales. Todas se basan en secuenciar clones desde ambos extremos. Estas estrategias incluyen: pair-ends, matepairs, BAC-ends, etc. Al secuenciar los fragmentos por ambos extremos se puede utilizar esta información en el ensamblaje, de modo que sabemos que esas dos lecturas van juntas y separadas por una distancia igual al tamaño del fragmento.
Terminología¶
Read: una secuencia obtenida del secuenciador.
Overlap: región alineada de dos o más secuencias.
Unitigs: ensamblajes (reads + overlaps) de copia única, suelen estar flanqueados por regiones repetidas.
Contigs: Unitigs indivuales o que solapan gracias a la información de paired ends o mate pairs.
Scalfolds: Contigs que no solapan entre si (tienen gaps) pero que se sabe que van juntos gracias a la información de los mate pairs.
Consense: Secuencia deducida a partir del ensamblaje de los reads.
Coverage: (Cobertura) Número de reads que cubren una base, es decir número de veces que se ha secuenciado una base. Suele referirse a la cobertura media.
N50: Es una medida de calidad del proyecto, se suele utilizar en proyectos de secuenciación de genomas. Para calcularlo se ordenan los contigs de mayor a menor, se suman por ese orden hasta alcanzar el 50% del tamaño del genoma, la longitud del último contig añadido es el valor N50. Es decir el tamaño de contig más pequeño que es necesario añadir para conseguir el 50% del genoma.
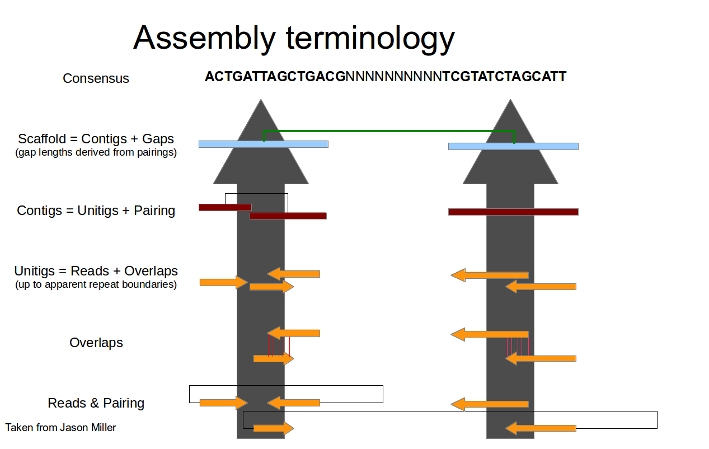
from Jason Miller
Programas¶
Se han desarrollado numerosos programas para el ensamblaje de secuencias tipo Sanger. Podemos destacar por ejemplo Phrap , Cap3 y el TIGR Assembler o su versión actualizada CABOG. Estos programas han sido diseñados y ampliamente utilizados para el ensamblaje de genomas. Existen otros paquetes de programas para proyectos de menor escala, nosotros vamos a utilizar en prácticas el Staden, que es un paquete de programas con GUI y de código libre. Está formado por una serie de programas como:
trev (editor de cromatogramas),
pregap (limpieza de secuencias) y
gap4 (ensamblador).
