Métodos predictivos en ADN y ARN¶
Una secuencia de nucleótidos no nos aporta ninguna información, salvo que seamos capaces de descifrarla. Para ello, debemos conocer el código de cifrado y tener las herramientas necesarias para descifrar la secuencia de una cadena de ADN. Usando estas herramientas podemos predecir sin datos experimentales que información contiene la secuencia. Podemos predecir la secuencia de la proteína que codifica, la localización de exones e intrones, etc. Pero hay que tener en cuenta que el resultado que obtenemos es una predicción teórica que deberá ser confirmada mediante experimentos.
La predicción de las estructuras presentes en una secuencia y el descifrado de la información son de vital importancia en los proyectos genoma; ya que nos enfrentamos a una secuencia que contiene numerosos genes que no han sido estudiados experimentalmente y de los cuales no se tiene ninguna información previa.
Traducción de una secuencia de nucleótidos¶
Traducir una secuencia en una de las predicciones más fáciles. Conocemos el código de la información y no presenta ninguna complicación el análisis.
El código es el código genético: cada aminoácido es codificado por unos varios tripletes y las señales de inicio y parada también están codificadas por tripletes. Lo único que tenemos que hacer es transcribir a ARN y leer la secuencia en formato triplete.
ADN AGGTTTACATGTAGAGGA.....TGA
RNA AGG UUU ACA UGU AGA GGA UGA
PROT Arg Phe Thr Cys Arg Ala Fin
El primer problema que nos planteamos es donde empezamos a traducir, ya que la traducción será diferente si comenzamos en el primer nucleótido, en el segundo o en el tercero.
ADN AGGTTTACATGTAGAGGA .....TGA
RNA1 AGG UUU ACA UGU AGA GGA......UGA
PROT Arg Phe Thr Cys Arg Ala Fin
RNA2 GGU UUA CAU GUA GAG GAU.....
PROT Gly Fin Leu Val Glu Asp.....
RNA3 GUU UAC AUG UAG AGG AUG....
PROT Val Tyr Met Fin Arg Met ...
Además también se puede traducir en el sentido inverso, es decir la cadena complementaria. Un fragmento de ADN tiene seis pautas de lecturas posible. En el emboss existe el programa transeq o sixpack.
Traducción de las seis pautas de un fragmento de ADN.

Los programas de traducción nos dan la secuencia de las proteínas codificadas por las pautas de lectura independientemente de que comiencen con un codón de inicio o contengan codones de parada. Únicamente leen el ADN.
Traducción inversa¶
También podemos deducir la secuencia de nucleótidos a partir de una secuencia de aminoácidos. El problema es que hay varios codones que pueden codificar el mismo aminoácido, por lo que la secuencia calculada tendrá ambigüedades. Programa backtranambig.
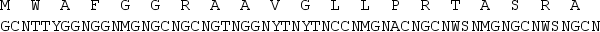
Búsqueda de pautas de lectura abierta (ORF)¶
En vez de traducir directamente la secuencia podemos hacer que el programa solo nos muestre aquellas regiones comprendidas entre un codón de inicio y un codón de parada. Además de mostrarlas hay programas que nos devuelven la secuencia de aminoácidos que codifica esa región. Por ejemplo el programa plotorf o showorf.

Búsqueda de dianas de restricción¶
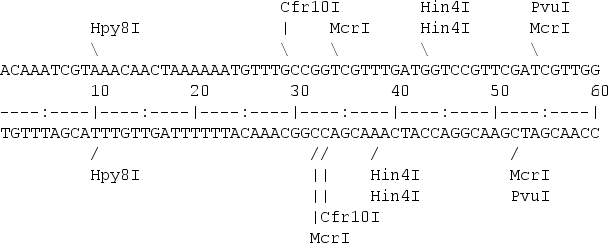
Estas herramientas localizan en la secuencia dianas de enzimas de restricción. Necesitamos una base de datos de las secuencias reconocidas por las diferentes enzimas, por ejemplo: Rebase. El programa nos localizará las dianas identificándolas en nuestra secuencia. Uno de los programas que podemos utilizar es remap .
Predicción de regiones promotoras¶
Hasta ahora nos hemos centrado en problemas poco complejos debido a que el código utilizado para almacenar la información es exacto y conocido (código genético, secuencias dianas). El reconocimiento de las regiones promotoras se basa en la identificación de las secuencias reconocidas por las proteínas generales del complejo de la transcripción y de los factores de transcripción específicos. Las dianas de los factores de transcripción están menos conservadas y presentan mucha variación en su secuencia, suelen ser secuencias muy cortas (tienen poca complejidad y existe bastante probabilidad de que las encontremos al azar) y además muchas de las dianas no están profundamente estudiadas con lo que partimos de información limitada. Esto complica la predicción de promotores, ya que no podemos centrarnos en la búsqueda de una secuencia exacta y además tendremos que valorar si la identificación es significativa o no tiene sentido biológico.
Secuencias de unión¶
Las secuencias que tenemos que reconocer pertenecen a dos tipos:
Secuencias presentes en la mayoría de promotores.
Secuencias en procariotas:
Pribnox box. Secuencia consenso TATAAT y situada a -10 pb del inicio de la transcripción.
TTGACA a -35 pb del inicio.
Secuencias menos conservadas en el inicio de la transcripción.
Secuencias en eucariotas:
TATA box. Secuencia TATAWDR (W A/T, D es no C, R es G o A) situada a -17pb del inicio.
CCAAT y GC box de secuencias y posición más variables.
Secuencias reconocidas por factores de transcripción específicos:
Cada factor de transcripción reconoce una determinada secuencia más o menos conservada.
No todos los genes están regulados por los mismos factores de transcripción.
La posición de las dianas es variable y pueden estar muy alejadas del inicio de la transcripción.
Debido a la falta de conservación de estas secuencias no las podemos definir con una secuencia exacta. Existen varios métodos para definir estas secuencias:
Secuencia consenso: Nos muestra para cada posición el nucleótido más abundante en las diferentes posiciones de las dianas conocidas para el factor de transcripción
TATAAT. Caja Pribnow. Pero no nos reconocería variantes existentes como TATAAA.
Patrones de secuencia. Nos muestran todas los posibles nucleótidos para cada posición.
TATAAW—TATAA(A/T). Nos reconoce todas las dianas conocidas pero no nos da
información sobre la veracidad de la secuencia. Si TATAAA está solo en el 2% de
los promotores, una TATAAA predicha es menos probable que una TATAAT que está en el
98%. Está información es más relevante cuando existen varias posiciones variables.
TWTAAW. Si cada posición variable se encuentra en un 1% la probabilidad de que la
secuencia TTTAAA sea una diana real es del 0,0001.
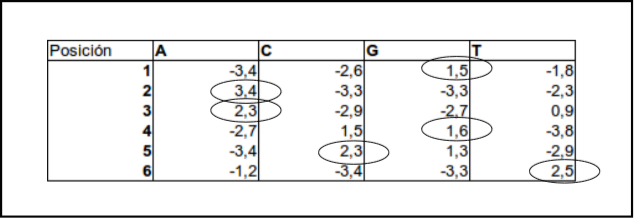
Matrices de puntuación. Nos da la puntuación de la existencia de cada nucleótido en cada posición.
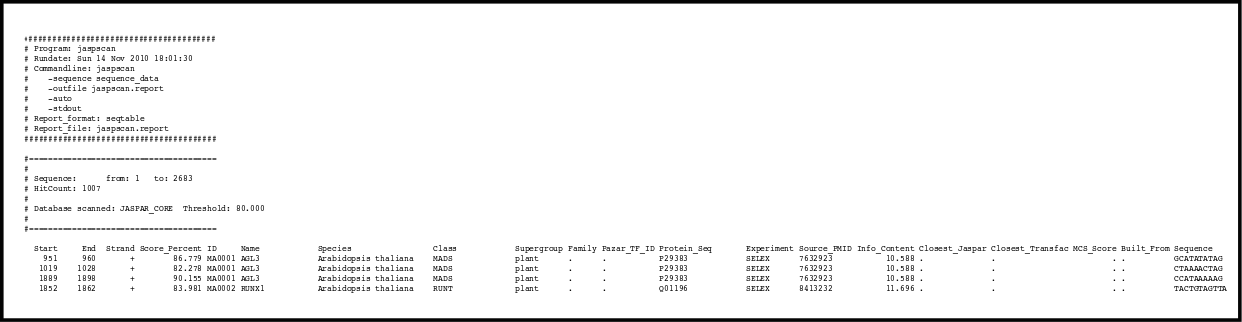
Existen diferentes métodos para identificar regiones promotoras, pero no existe ninguno que sea capaz de predecir con exactitud estas regiones. La mayoría de ellos reconoce entre 16-58% de los promotores; pero comete muchas falsas identificaciones. A su vez se han desarrollado numerosos programas para la identificación de promotores y factores de transcripción, algunos ejemplos: Tfscan , Jaspscan o GrailEXP.
Predicción de pautas de lectura¶
La ventaja al trabajar con procariotas es que sus genes no están separados por intrones y que sus regiones intergénicas no son grandes y al igual que en los mRNAs eucarióticos las regiones codificantes no están fragmentadas. En teoría la predicción de regiones codificantes correspondería con la identificación de los genes, ya que estos contienen una pauta de lectura continua. En caso de genomas eucarióticos la predicción de regiones codificantes tiene que ir acompañada de la predicción de la estructura exón e intron, al estar las pautas truncadas.
El primer problema lo encontramos al buscar pautas abiertas de lectura. Para un mismo fragmento podemos encontrar una ORF en calquiera de las pautas de lectura.
Búsqueda de pautas de lectura abiertas
Para identificar la pauta de lectura se puede utilizar diferentes métodos. Uno de ellos está basado en la predilección de codones de cada especie. Existen varios codones que codifican para el mismo aminoácido; pero no todos los organismos los utilizan en la misma proporción. Con estos datos podemos construir una matriz de pesos de uso de codones que podemos utilizar para valorar cada ORF encontrada.
Tabla de uso de codones en Drosophila melanogaster
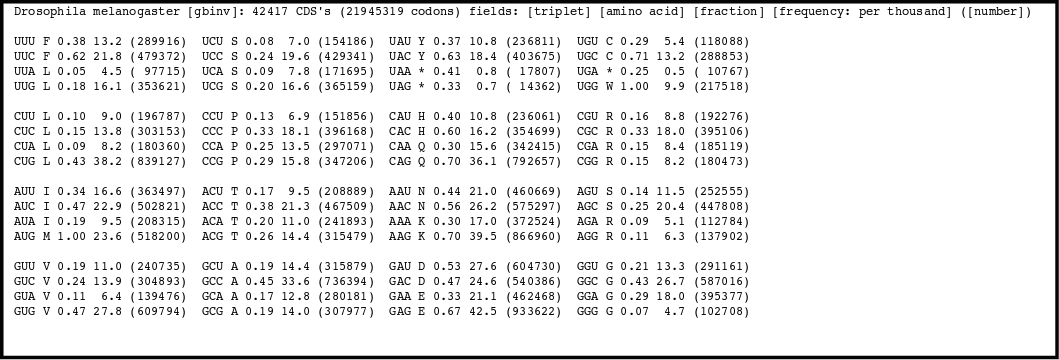
from http://www.kazusa.or.jp/codon
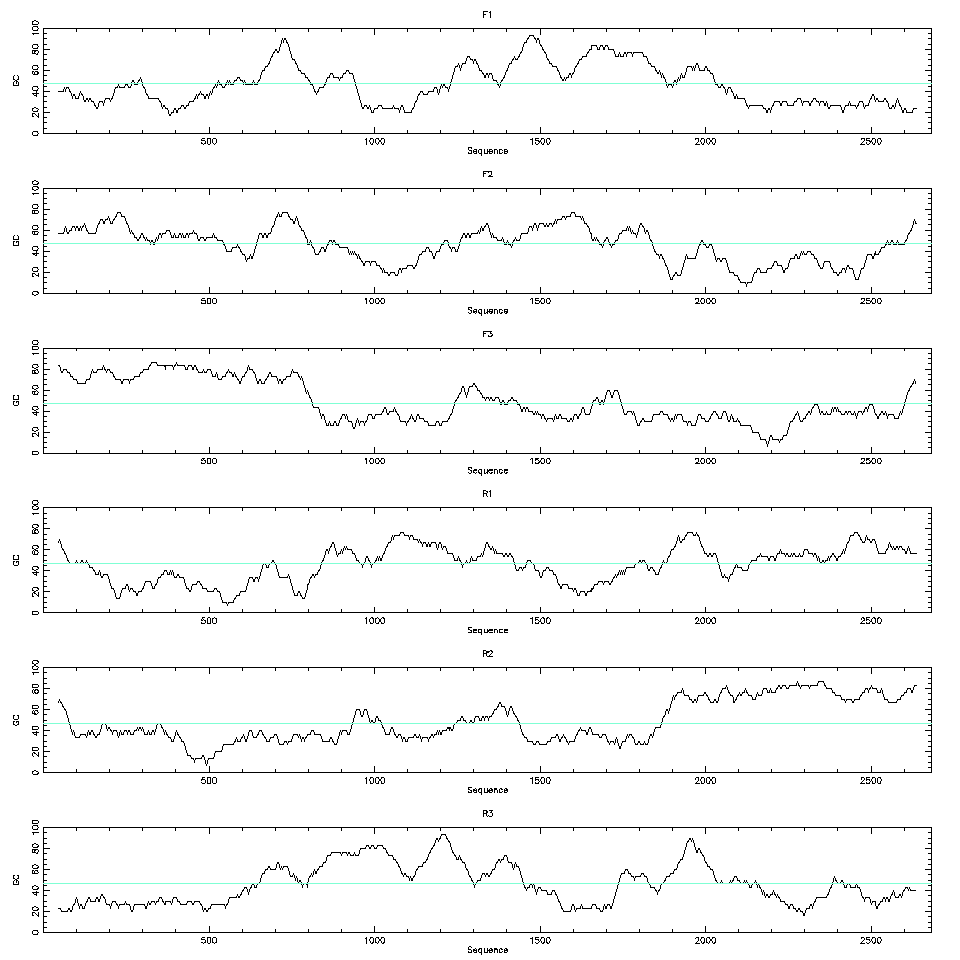
Otro sistema es en que la frecuencia de uso de los nucleótidos en la tercera base del codón no es aleatoria, tendiéndose a usar la misma base. Esto es debido a que los codones no son usados al azar. A diferencia del método anterior no es necesario tener una matriz de uso de codones; por lo que lo podemos aplicar a especies con pocas secuencias. Existen programas que nos proporcionaran un gráfico de la no distribución al azar de esta posición frente a la distribución al azar en las regiones no codificantes. Por ejemplo podemos utilizar el programa wobble .
Porcentaje de GC en la tercera posición del codón de un gen de Drosophila melanogaster
El último sistema se basa en la búsqueda de secuencias homólogas en las bases de datos. Podemos traducir todas las ORF y comprobar si alguna de ellas presenta alta similitud con proteínas conocidas.
Estos tres sistemas se basan en la identificación correcta de las ORF, nos van a ayudar a discernir cual de ellas tiene mayor probabilidad de ser la expresada en la realidad. Pero esto lleva una serie de problemas. La mayoría de ellos relacionados con la inexactitud de las secuencias, podemos tener errores en la secuencia que provoquen cambios de un codón a otro y inserciones o delecciones que rompan la pauta de lectura. En estos casos este abordaje nos llevaría a errores en la determinación de las ORFs.
Existen estrategias para solucionar este problema. Una de ellas esta basado en el uso de los codones y tercera base no es al azar. El programa va analizar hexámeros de nucleótidos de regiones ya caracterizadas para determinar la probabilidad de asociaciones de estos nucleótidos en regiones codificantes y no codificantes. Con ello se crea un modelo de región codificante que será especifico para la especie analizada. Este modelo permite analizar y detectar las regiones con mayor probabilidad de ser codificantes y no se verá tan afectado por mutaciones puntuales o cambios de pauta de lectura. Un programa que utiliza este sistema es el Genemark_HMM .
Predicción de la estructura génica¶
Predecir la estructura de un gen en procariotas es posible utilizando la combinación de los dos análisis que hemos visto: detección de regiones promotoras y búsqueda de ORFs. Pero en eucariotas la situación es mucho más compleja, los genes presentan intrones y exones (tienen la pauta de lectura fragmentada), hay exones que no presentan regiones codificates, el tamaño de las regiones no codificantes (intrones , regiones intragénicas, promotores, etc) pueden ser muy grandes.
Búsqueda de regiones codificantes¶
La podemos realizar por los métodos vistos anteriormente, localizando pautas de lectura abiertas y validándolas con los métodos ya comentados (uso de codones, tercer posición, o métodos más complejos como los del Genemark). Otro modo es buscar homologías usando la traducción de las seis pautas de lectura, con blastx por ejemplo.
Estos métodos no resultan muy prácticos cuando nos enfrentamos a genes fragmentados, aunque son útiles para buscar las ORFs en cDNAs o genes sin intrones
Comparación con mRNAs o EST¶
Una forma de conseguir la correcta ORF es determinar las regiones exónicas e intrónicas de modo que podamos recomponerla juntando los exones. Existen varios métodos para determinar la estructura de un gen.
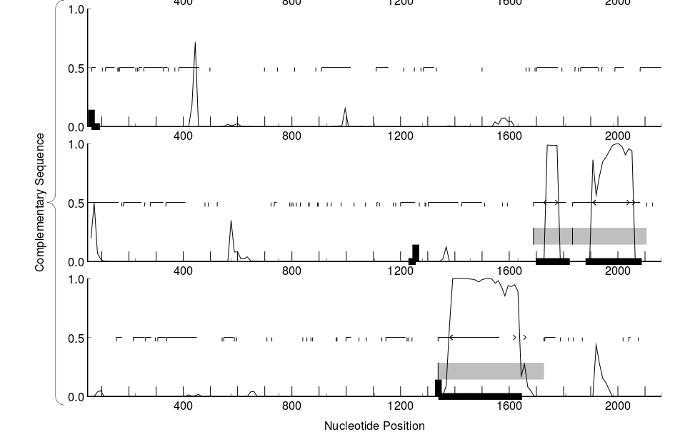
Uno forma de encontrar la estructura es alinear las secuencias genómicas con los mRNAs o secuencias de los ESTs disponibles. De este modo determinaremos exactamente las regiones exónicas. En el emboss podemos utilizar: esim4 (mRNAs) o est2genome (ESTs).
Alineamiento de un EST con un fragmento genómico
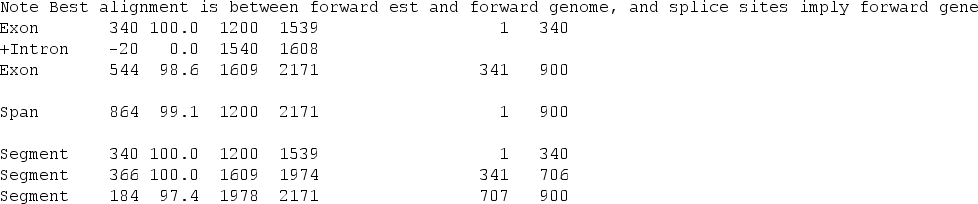
Este método tiene el inconveniente de que necesitamos tener ESTs o mRNAs de los genes y esto no es siempre posible. De hecho muchos de los genes no los vamos a tener representados en las colecciones de ESTs. Otro problema es que el gen no este cubierto totalmente por los ESTs, lo que nos produciría una identificación parcial de la secuencia.
Uso de redes neuronales y búsqueda de patrones¶
Existen diferentes programas que van a combinar diferentes análisis para identificar genes y predecir su estructura. El método de abordaje es diferente (redes neuronales o búsqueda de patrones) pero se basan en la identificación y puntuación de características como por ejemplo:
Analizar hexámeros de nucleótidos de regiones ya caracterizadas para determinar la probabilidad de asociaciones de estos nucleótidos en regiones codificantes y no codificantes en las diferentes pautas de lectura.
Analizar porcentaje de GC de regiones ya caracterizadas para determinar la probabilidad de presencia de zonas ricas en GC en regiones codificantes y no codificantes.
Búsqueda de sitios dadores y aceptores de splicing.
Presencia de pautas de lectura abiertas.
Ya sea con el uso de redes neuronales o búsqueda de patrones estos sistemas son calibrados y evaluados para una determinada especie. No podemos utilizar la calibración para identificar otra especie para la cual no han sido entrenados. El porcentaje de acierto depende del programa utilizado, del entrenamiento al que se ha sometido y de la complejidad de las estructuras. Programas muy utilizados son el Genemark_HMM, el GeneScan o Augustus.
Predicción de Genemark HMM
Es necesario combinar diferentes estrategias de búsqueda de regiones codificanes y predicción de estructuras para poder determinar con precision las ORF de los genomas eucariotas.
