SECUENCIACIÓN DE SANGER¶
Secuenciación mediante Sanger¶
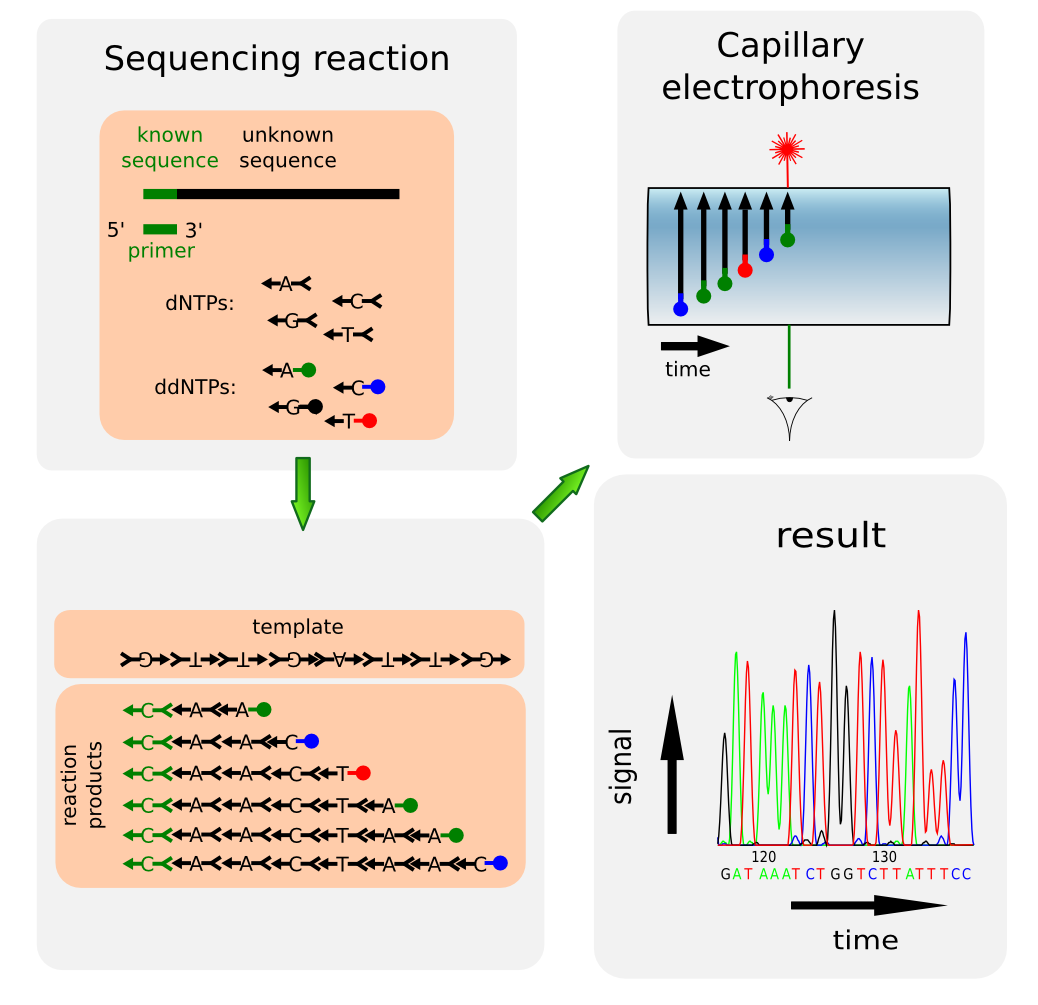
La secuenciación de Sanger se basa en la polimerización del ADN y el uso de dideoxinucleótidos que sirven como terminadores de la reacción. En la actualidad la reacción de secuenciación se basa en una modificación de la PCR con dideoxinucleótidos marcados con fluoróforos y se resuelve mediante una electroforesis capilar. El sistema más utilizado es el desarrollado por Applied Biosystems . Podéis encontrar numerosos documentos sobre la secuenciación de Sanger, pero para comenzar esta entrada de la wikipedia .
Cromatograma.
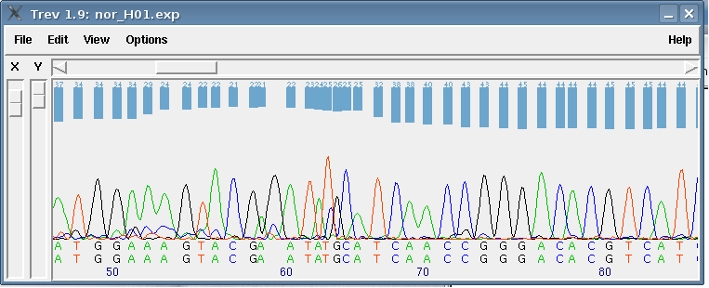
Cromatogramas¶
La información obtenida en los secuenciadores automáticos se guarda en ficheros binarios.
Estos ficheros pueden incluir, además del cromatograma procesado (trace), el dato crudo leído por el secuenciador automático, la secuencia de nucleótidos y las calidades.
Hasta el momento el mercado de la secuenciación automática basada en el método de Sanger está controlado principalmente por Applied Biosystems.
Los secuenciadores de Applied generan los datos en formato abi.
Estos ficheros abi se pueden leer con distintos programas: chromas (Win), el Sequence Scanner (Win) o el trev del paquete de análisis Staden (Mac, Pc, Linux).
Otro formato en el que se guardan los cromatogramas es el scf.
scf es un formato libre no controlado por una empresa.
scf sólo incluye el cromatograma procesado y la secuencia.
En el NCBI se depositan algunos de los cromatogramas obtenidos en los proyectos de secuenciación.
Basecalling¶
A partir de los cromatogramas hay que obtener la secuencia de nucleótidos.
Este proceso lo hacen automáticamente los programas que leen los cromatogramas, pero conviene que revisarlo manualmente porque en muchas ocasiones se producen fallos al asignar las bases.
La secuencia propuesta por el software del secuenciador automático casi siempre tiene una gran parte al final que hay que eliminar.
Calidad¶
Todos los sistemas de secuenciación estiman la probabilidad de que cada uno de los nucleótidos secuenciados sean erróneos, a este parámetro se le suele denominar calidad. Esta estimación del error es específica de cada tecnología y la calcula el software del equipo. Para facilitar el análisis y la interpretación de los resultados, estos valores se suelen cambiar a una escala normalizada que utilizan todas las tecnologías de secuenciación, la escala de Phred. Phred era originalmente un programa de basecalling, pero ahora es sobretodo conocido como la escala del valor de calidad. Está se define como:
Phred score = - 10 log (prob error).
De este modo es fácil interpretar la probabilidad de error según su valor.
Phred score
Probabilidad de error de lectura en esa base
10
1/10
20
1/100
30
1/1000
40
1/10000
Interpretación de los cromatogramas de secuencia¶
Ejemplos obtenidos de la web del servicio de secuenciación de la Universidad de Michigan.
Ruido¶
En teoría un cromatograma siempre debería ser perfecto, pero no siempre es así.
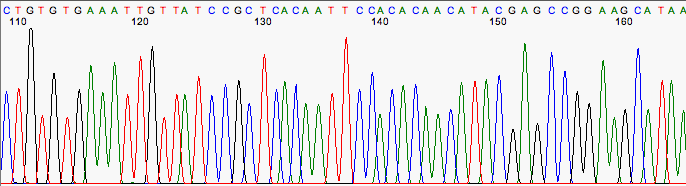
Un buen cromatograma.
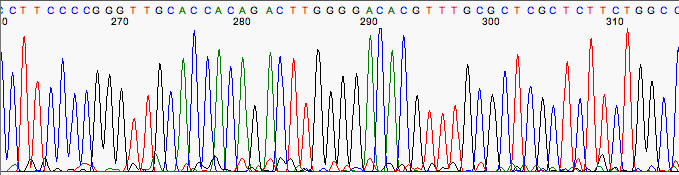
Cromatograma con algo de ruido.
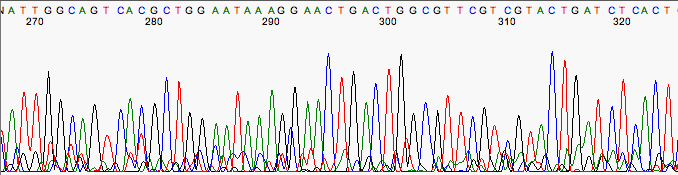
Cromatograma con mucho ruido.
Posibles causas del ruido de fondo son: una contaminación de otro ADN o una contaminación con otro cebador (normalmente el cebador reverso utilizado en la PCR).
Errores en el basecalling¶
Algunas veces el secuenciador automático no es capaz de colocar los picos correspondientes a las distintas bases a una distancia equidistante. Por ejemplo esto sucede con frecuencia en el dinucleótido GA.
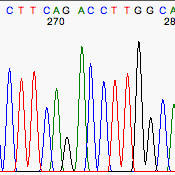
Mal espaciado bien interpretado.
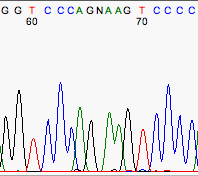
Mal espaciado que introduce una N extra.
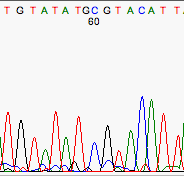
Mal espaciado y fondo que introduce una base extra.
Heterocigotos¶
Los programas de basecalling suelen interpretar los heterocigotos como N.
Hay programas especializados en detectar estos picos dobles y en etiquetarlos adecuadamente.
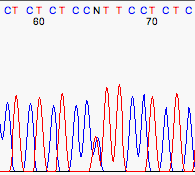
Heterocigoto C/T.
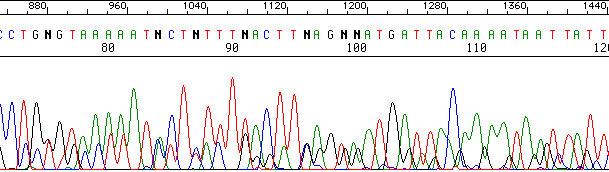
Pérdida de resolución¶
Incluso las buenas secuencias pierden resolución al avanzar la secuencia, debido a la cromatografía.
Este es uno de los motivos que hacen que las lecturas no tenga más de 700-800 pb.
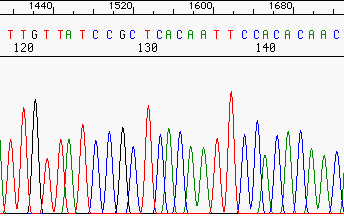
Resolución buena.
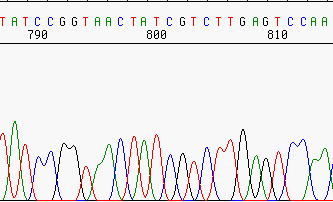
Resolución aceptable.
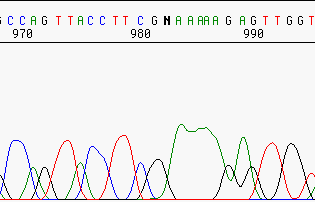
Resolución mala.
Problemas durante la reacción de secuenciación¶
En algunas ocasiones pueden haber problemas en la reacción de secuenciación que impiden tener una buena secuencia.
Es necesario diagnosticar el problema para solucionarlo.
Posibles causas para los problemas en las secuencias.
No hay señal:
No había ADN molde.
No había cebador.
El cebador no ha reconocido al molde.
La señal es muy débil:
Había poco ADN molde.
El cebador no ha reconocido bien al molde.
Hay señal, pero la resolución es mala desde el principio:
Puede que haya un contaminante en la muestra que afecta a la cromatografía.
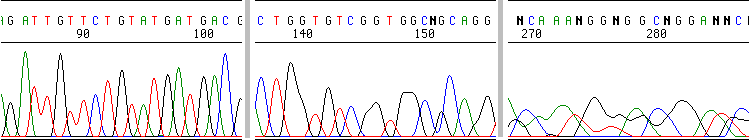
La señal y la resolución son buenas, pero hay varias bandas en cada posición:
Varios moldes en la reacción:
El cebador se une en varias posiciones del molde.
Se están secuenciando varios productos.
Los cebadores de la PCR original no se han eliminado.
Pérdida gradual de señal en los tamaños grandes:
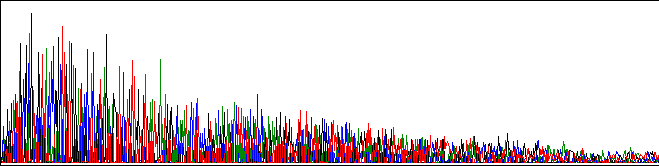
La señal es buena al principio, pero disminuye rápidamente:
Exceso de sales en el molde.
Exceso de ADN molde.
Contaminante que inhibe a la polimerasa.
Un gran pico rompe la secuencia en un punto concreto.
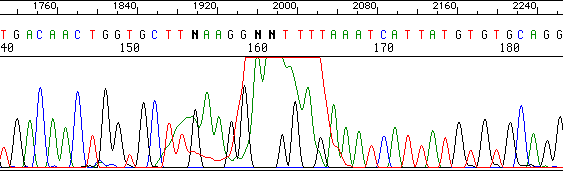
ddNTPs mal eliminados de la reacción de secuenciación.
La señal es buena hasta un punto concreto y después disminuye bruscamente:
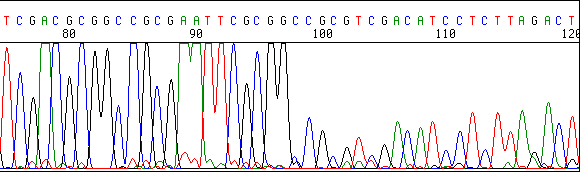
Estructura secundaria en el ADN molde.
Visualización de cromatogramas¶
Descargar los el fichero cromprimido de cromatogramas de ejemplo.
Abrir los cromatogramas que habíamos descargado con el trev de uno en uno y comprobar su calidad.
Conviene pedir al trev que marque la calidad de la asignación de cada base (view -> display confidence).
Marcar las regiones de mala calidad del principio y el final de las secuencias (edit -> left quality, edit -> right quality).
Evaluar para cada un de ellos:
¿Tiene señal? ¿Se ven bandas o es todo ruido?
¿Dónde comienza la región de buena calidad? ¿Dónde termina?
¿Acaba la región de mala calidad de forma brusca?
Posible diagnóstico de los problemas.
¿Hay bases mal interpretadas por el basecaller?
¿Hay mucho ruido de fondo?
Guardar la secuencia obtenida en texto plano.
